Background: Retinitis pigmentosa is the most common retinal dystrophy (RP), and it can be non-heritable, heritable, or in association with systemic disorders. There is variability between presentation, which correlates with inheritance pattern. Autosomal recessive has severe vision loss and night blinds that occurs early in life. Autosomal dominant has a more gradual onset that occurs in adult life, with variable penetrate and less severe vision loss. X-linked recessive is the rarest form and most severe, with onset similar to autosomal recessive. A salt-and-pepper fundus can be seen in females in the X-linked variant. Sporadic nature is also a possibility with RP. RP is most commonly autosomal dominant, and Usher’s syndrome is the most common associated systemic condition associated with RP. Usher’s syndrome is an autosomal recessive condition that is recognized with congenital hearing loss. Additionally, Usher’s syndrome accounts for half of the blind and dead individuals. Retinitis pigmentosa is most commonly diagnosed at age 9-19. There is no race predilection for this retinal dystrophy. (2)
Systemic Associations: Numerous syndromes that feature RP can be seen, along with systemic abnormalities. These associations are normally metabolic disorders seen with atypical forms of RP. A careful review of symptoms is important and critical in RP cases. (2)
Pathophysiology: RP is a generic term that classifies a group of hereditary conditions (29 loci associated with various phenotypes); additionally, it is characterized by progressive and gradual loss of RPE and photoreceptor function. Rod damage is more prominent than cone damage. (3)
Symptoms: Patients will notice peripheral vision loss and night blindness. Night blindness is the most common symptom, and peripheral vision loss is noted in early stages while in dim light. Decreased central vision can occur early or late in the progression of RP, and color vision is typically intact until advanced and late stages. Symptoms usually are gradual over many years, with approximately 75% of patients with RP showing symptoms by the age of thirty. (4)
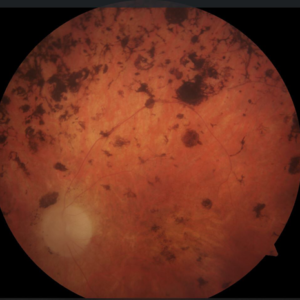
Classic triad: The classic triad for retinitis pigmentosa is vessel attenuation, bone-spicule pigmentation, and waxy optic disc pallor. (4)
Other signs: Posterior subcapsular cataracts, cystoid macular edema within the outer plexiform layer of the retina, and severe optic nerve head disc drusen can additionally be noted. Epiretinal membranes, macular atropy, keratoconus, myopia, and progressive narrowing of peripheral visual field (such as a ring scotoma that progresses to a small central visual field) can also be seen. These patients will also have prolonged dark adaptation. (4)
Atypical Presentations: Atypical presentations may represent either incomplete forms of the disease or closely related conditions. RP sine pigmento is when pigmentary changes are initially absent or minimal. Sector RP is when pigmentary changes are only in one quadrant or two. This pigment is often just in one sector near the posterior pole. Retinitis punctata albescens is scattered whitish spots, located mostly near the equator. (2)
ERG Testing: An electroretinogram measures electrical potentials and records graded potentials produced within the retina in response to light. The outer retinal layers, such as layers that include photoreceptor cells and bipolar cells, are examined in an ERG. Prior to performing an ERG, the patient is dark adapted for approximately forty-five minutes. Next, the retina is flooded with various light intensities and wavelengths. The patient is tested under dark conditions to test rod function as well as light conditions to test cone function. In the early stages of RP, only the scotopic, or rod, ERG is abnormal. In the late stages of RP, the entire ERG is entirely extinguished due to poor rod and cone function. (3)
Treatment: There is currently no cure for RP. However, the use of vitamin A supplementation may slow down the progression. Adults receive 15,000 IU/day of vitamin A (palmitate form), and vitamin A consumption in children is controversial. The recommended dosage is 5,000 IU/day for six year olds and 10,000 IU/day for ten year olds. A pediatrician should be consulted before use of vitamin A in children to weight risk and benefit. If vitamin A therapy is instilled in either adults or children, liver function tests and vitamin A levels should be closely monitored. It should be noted that vitamin E should never be used in a patient with RP, as vitamin E worsens this retinal dystrophy. Patients with functional vision loss can be treated with low vision aids, tints, and visual field enhancers. The tinted lenses will provide comfort outdoors as well an enhanced contrast. If a patient has cystoid macular degeneration, topical or oral carbonic anhydrase inhibitors may be effective. Cataract extraction in patients with advanced cataracts can also potentially improve acuity. Additionally, all patients benefit from genetic counseling and instruction on how to deal with visual handicaps stemming from RP. In late stages of advanced dystrophy, vocational rehabilitation can be beneficial. Epiretinal and subretinal microchip implants are another potential option that can be explored in patients with advanced RP. Ongoing clinical trials are evaluating the safety and efficacy of retinal implants. In addition, gene therapy research is ongoing but not yet clinically available. (1)
References:
- Bagheri, N., & Wajda, B. (2017).The Wills Eye Manual: Office and Emergency Room Diagnosis and Treatment of Eye Disease (Seventh ed.). Philadelphia: Wolters Kluwer.
- Bowling, B., & Kanski, J. J. (2016). Kanski’s Clinical Ophthalmology : a Systematic Approach (8th ed.). Edinburgh: Elsevier.
- Cheatham, K. M., Cheatham, M. A., Wood, K. B., & Kmk Educational Services, Llc. (2007). KMK Part One : Basic Science Review Guide (12th ed.). Overland Park, KS: Kmk Educational Services, Llc.
- Cheatham, K. M., & Wood, S. D. (2018).KMK Part Two: Diagnosis and Treatment: Board Review Guide (7th ed.). Omaha, Nebraska: KMK Educational Services, LLC.